当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Revealing Carbon Vacancy Distribution on α-MoC1–x Surfaces by Machine-Learning Force-Field-Aided Cluster Expansion Approach
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-06-30 , DOI: 10.1021/acs.jpcc.3c01941
Jun-Zhong Xie 1 , Hong Jiang 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2023-06-30 , DOI: 10.1021/acs.jpcc.3c01941
Jun-Zhong Xie 1 , Hong Jiang 1
Affiliation
|
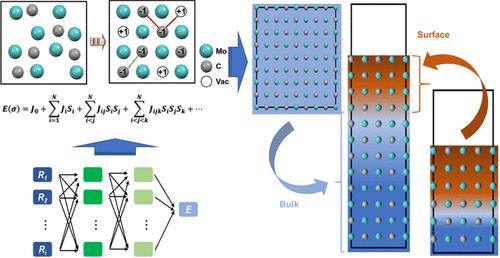
α-MoC1–x has recently attracted extensive attention in heterogeneous catalysis for its unique catalytic properties. As a nonstoichiometric material, the performance of α-MoC1–x is closely related to its intrinsic carbon vacancy, but the knowledge regarding the structural nature associated with the vacancy is still lacking. In this work, we perform a cluster expansion (CE) study to reveal the vacancy distribution on α-MoC1–x (001) and (111) surface. Considering that a large number of symmetrically distinct clusters are needed to build accurate CE models for multicomponent surface systems, we adopt the strategy of utilizing the machine-learning force field to efficiently generate thousands of training data. With the effective cluster interactions on the α-MoC1–x (001) and (111) surfaces, Monte Carlo simulations are conducted to model the surface vacancy distribution behaviors. The surface-bulk difference as well as the surface effects on configurational properties are revealed. It is found that the carbon atom tends to segregate to the topmost layer of the α-MoC1–x (001) surface and the vacancy tends to segregate to the topmost layer of the α-MoC1–x (111) surface. We also predict the structures of the topmost layer of (001) and (111) surface under the simulated reaction condition, which can provide important insights into catalyst modeling and design for α-MoC1–x-related catalytic systems. The scheme to build surface CE models developed in this work can be generally used in theoretical modeling of heterogeneous catalytic surfaces with configurational disorder.
中文翻译:

通过机器学习力场辅助簇扩展方法揭示 α-MoC1–x 表面上的碳空位分布
α-MoC 1– x因其独特的催化性能近年来在多相催化领域引起了广泛关注。作为一种非化学计量材料,α-MoC 1– x的性能与其固有的碳空位密切相关,但与空位相关的结构性质的知识仍然缺乏。在这项工作中,我们进行了簇扩展(CE)研究,以揭示 α-MoC 1– x上的空位分布(001)和(111)表面。考虑到需要大量对称不同的簇来为多组分表面系统建立精确的CE模型,我们采用利用机器学习力场的策略来有效地生成数千个训练数据。利用 α-MoC 1– x (001) 和 (111) 表面上的有效团簇相互作用,进行蒙特卡罗模拟来模拟表面空位分布行为。揭示了表面-体积差异以及表面对构型特性的影响。研究发现碳原子倾向于偏析到α-MoC 1– x (001)表面的最上层,空位也倾向于偏析到α-MoC 1– x的最上层(111)表面。我们还预测了模拟反应条件下(001)和(111)表面最顶层的结构,这可以为α-MoC 1– x 相关催化系统的催化剂建模和设计提供重要的见解。本工作中开发的建立表面CE模型的方案可普遍用于具有构型无序的异质催化表面的理论建模。
更新日期:2023-06-30
中文翻译:
通过机器学习力场辅助簇扩展方法揭示 α-MoC1–x 表面上的碳空位分布
α-MoC 1– x因其独特的催化性能近年来在多相催化领域引起了广泛关注。作为一种非化学计量材料,α-MoC 1– x的性能与其固有的碳空位密切相关,但与空位相关的结构性质的知识仍然缺乏。在这项工作中,我们进行了簇扩展(CE)研究,以揭示 α-MoC 1– x上的空位分布(001)和(111)表面。考虑到需要大量对称不同的簇来为多组分表面系统建立精确的CE模型,我们采用利用机器学习力场的策略来有效地生成数千个训练数据。利用 α-MoC 1– x (001) 和 (111) 表面上的有效团簇相互作用,进行蒙特卡罗模拟来模拟表面空位分布行为。揭示了表面-体积差异以及表面对构型特性的影响。研究发现碳原子倾向于偏析到α-MoC 1– x (001)表面的最上层,空位也倾向于偏析到α-MoC 1– x的最上层(111)表面。我们还预测了模拟反应条件下(001)和(111)表面最顶层的结构,这可以为α-MoC 1– x 相关催化系统的催化剂建模和设计提供重要的见解。本工作中开发的建立表面CE模型的方案可普遍用于具有构型无序的异质催化表面的理论建模。
































 京公网安备 11010802027423号
京公网安备 11010802027423号